
BRAF靶点抑制剂中国临床研究现状
 2023-07-01
2023-07-01 
 MCE
MCE
1.靶点机制
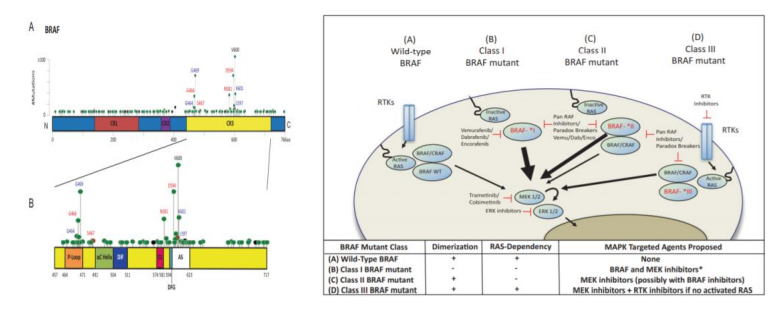
BRAF 基因位于染色体 7q34,编码丝氨酸/苏氨酸蛋白激酶,是 RAF 家族成员。BRAF蛋白与 KRAS 蛋白同为 RAS-RAF-MEK-ERK 信号通路中上游调节因子,使 MEK 蛋白磷酸化,随后 ERK 蛋白磷酸化,系激活参与细胞增殖和生存的相关基因。突变的 BRAF 蛋白增强了激酶的活性,可在体外转化。而其中具有致癌以及治疗价值的是 V600 突变,主要包括 V600E 和 V600K 突变。该位点的突变可引起下游活化致癌,占整体 BRAF 突变的一半。大多数 BRAF 突变的患者既往有吸烟史,病理类型是腺癌。BRAF 突变一般与 EGFR、KRAS 等突变相互独立和排斥,并不同时出现。
按照作用靶点的不同,BRAF 抑制剂分为多靶点激酶抑制剂和 BRAF V600E(单靶点)抑制剂两类。多靶点激酶抑制剂,如索拉非尼(Sorafenib) 、瑞戈非尼(Regorafenib) 、培唑帕尼(Pazopanib)、ASN-003 和 CEP-32496 等(详见多靶点 TKI 部分),对于包括 BRAF 在内的多种激酶均有抑制作用,具有广谱的抗肿瘤及抗血管生成作用,属于非特异性 BRAF抑制剂。特异性 BRAF V600E(单靶点)抑制剂,如维莫非尼(Vemurafenib)、达拉菲尼(Dabrafenib)、PLX-8394 和康奈非尼(Encorafenib)等,对 BRAF 尤其是 BRAF V600E 有很高的抑制活性,目前主要获批用于治疗黑色素瘤。
2.中国临床研究申报现状
目前国内进行中的 BRAF 抑制剂临床研究共 20 余项。其中Ⅱ期临床研究有 4 项,比如达拉非尼联合曲美替尼治疗 BRAF 突变阳性肺癌的研究,DCC-2618 在胃肠道间质瘤患者中的有效性、安全性及 PK 特征的研究, Encorafenib 联合西妥昔单抗治疗 BRAF 突变晚期肠癌的研究等。目前Ⅰ期临床研究共 5 项,如 RX208 在晚期恶性实体肿瘤患者中的Ⅰ期临床研究,TQ-B3233 胶囊Ⅰ期耐受性和药代动力学临床试验,评价单次和重复口服给药的药代动力学和安全性,还有评价 GSK2118436 单药和联合给药的药代动力学和安全性等。
资料来源:DANKNER M, ROSE AAN, RAJKUMAR S,et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations[J]. Oncogene,2018,37(24):3183-3199. doi:10.1038/s41388-018-0171-x. Epub 2018 Mar 15. PMID: 29540830.
3.简评
BRAF 蛋白是 MAPK/ERK 信号通路中重要的上游调节因子,其 V600E 突变可激活下游 MEK 蛋白,进一步引起肿瘤细胞生长、增殖和侵袭,易发生于结直肠癌、甲状腺癌、黑色素瘤等多个癌种之中,该类患者往往预后较差。虽然第一代 BRAF 抑制剂(Vemurafenib 和 Dabrafenib)在 BRAF V600E 突变的黑色素瘤患者上取得了良好的效果,但是用于其他肿瘤患者的 BRAF V600E 突变(如结直肠肿瘤)效果却不佳,且在一年之内均产生抗药性。因此,BRAF V600E 抑制剂往往要联合 EGFR 单抗或 MEK 抑制剂,才能够有效改善该类患者的生存现状及预后(如 Encorafenib 与西妥昔单抗或 Binimetinib 组合疗法)。新一代的 BRAF 抑制剂(如 PLX-8394、CEP-32496 等),也成为现阶段的研发方向之一。BRAF 作为抗肿瘤治疗中一个热门的分子靶点,尽管目前已有 20 多项临床研究正在进行中,但是其中Ⅱ、Ⅲ期研究仍然较少,高效的靶点抑制剂值得进一步探索。

